Primary malignant cardiac tumors are very rare. Rhabdomyosarcoma is the most common malignant cardiac tumors. These tumors usually arise from the left atrial walls. We presented a case of recurrence of primary cardiac rhabdomyosarcoma in a 35 year old woman presented as superior vena cava syndrome after 20 months of resection. The tumor arose from the posterior wall of the right atrium and extended to the tricuspid valve leaflet. Histopathology confirmed recurrence of the cardiac rhabdomyosarcoma.
Rhabdomyosarcoma; Recurrence
Primary malignant cardiac tumors are very rare with the incidence between 0.0017% to 0.28%. Most of them are sarcomas occurring in humans irrespective of sex and age, mainly during the third and fourth decades of life. They are most frequently found in the right and/or left atrium, and less frequently in the right or left ventricle [1].
Rhabdomyosarcoma is a malignant soft tissue sarcoma thought to arise from primitive mesenchymal cells that typically differentiate into skeletal muscle tissue. It is the most common type of soft tissue sarcoma in children and adolescents, with an annual incidence of 1 in 170,000 [1].
Rhabdomyosarcoma carries a high risk of local recurrence and regional lymph node spread, while the risk of distant metastasis is relatively lower. There are two main histological subtypes: embryonal (about 80% of cases) and alveolar (15–20% of cases).
The prognosis of cardiac Rhabdomyosarcoma is generally very poor; the survival time is reported to be from several weeks to two years [2].
Most cases of rhabdomyosarcoma are sporadic, but familial syndromes may also be associated. These malignant tumors are capable of invading the myocardium and extending into cardiac chambers. In our case, a diagnosis of rhabdomyosarcoma was made for a mass located in the right atrium.
Cardiac rhabdomyosarcoma is a rare and very aggressive malignant neoplasm. Most cases are primary tumors with poor outcomes despite the combination of surgery, chemotherapy, and radiotherapy [3].
A 35-year-old female patient who previously underwent surgery for 20 months at an external center for a right atrial rhabdomyosarcoma presented to our clinic due to swelling in the neck and face, dyspnea and palpitations. Upon follow-up, a recurrent right atrial mass was detected, and the patient was referred to our institution for surgical management
Physical Examination
Blood pressure: 120/60 mmHg, Heart rate: 110 bpm. The patient was alert, oriented, and cooperative. Pupils were isochoric. No carotid bruits were auscultated. Jugular venous distension was present. Thyroid was non-palpable. No lymphadenopathy was detected. Respiratory examination revealed symmetrical chest expansion and normal bronchovesicular breath sounds. Cardiac auscultation showed 3/5 holosystolic murmurs at the right sternal edage. hepatosplenomegaly was observed. Peripheral pulses were palpable. Neurological examination revealed no deficits. Other systemic examinations were unremarkable.
TEE A large mass was identified within the right atrium. TEE revealed a 5.6 × 4.4 cm mass with markedly irregular (malignant) borders. A portion of the mass extended into the tricuspid valve during systole, causing moderate-to-severe tricuspid regurgitation.
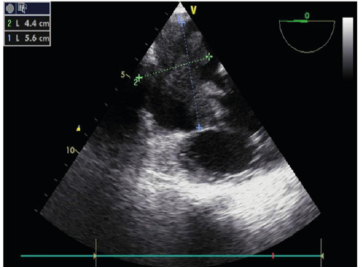
Figure 1: TEE revealed a 5.6 × 4.4 cm mass with markedly irregular borders
Thoracic Aorta CT Angiography
Cardiac enlargement was observed. The right atrium was dilated, and a lobulated mass measuring 62 × 40 × 46 mm was detected within the right atrium. Vascular structures were noted at the superior aspect of the mass. The lesion extended slightly through the tricuspid valve into the right ventricle and minimally toward the inferior vena cava.
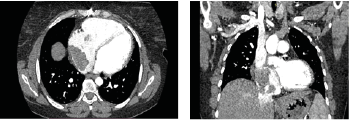
Figure 2 A and 2B: The right atrium was dilated, and a lobulated mass measuring 62 × 40 × 46 mm was detected within the right atrium.
Given the family history suggesting rhabdomyosarcoma in the patient’s siblings, the patient underwent surgery. With cardiopulmonary bypass support, the right atrial tumor was resected. The intraoperative period was uneventful, and the patient was transferred to the cardiovascular intensive care unit under inotropic support.
Histopathology following radiotherapy and chemotherapy showed a mesenchymal tumor with an infiltrative growth pattern, containing hemorrhagic and necrotic areas. The tumor consisted of fascicular and solid patterns of round to oval cells with eccentrically located nuclei, significant pleomorphism, and atypia.
Postoperative Course
On postoperative day 0, the patient was managed in the intensive care unit under dopamine and norepinephrine support. Her rhythm fluctuated between atrial fibrillation and sinus tachycardia, prompting amiodarone loading followed by maintenance dosing. Due to hypotension, the dose of adrenaline was increased and a furosemide infusion was started. Metabolic acidosis was observed on arterial blood gas analysis, and right-sided heart failure was suspected. Milrinone infusion was initiated.
On postoperative day 1, the patient remained intubated with poor diuresis and inadequate urine output. Routine labs revealed creatinine 1.21 mg/dL, ALT 618 U/L, and AST 698 U/L, raising suspicion for multi-organ failure. Hemofiltration was planned, and a femoral dialysis catheter was placed. However, the patient developed sudden ventricular fibrillation, CPR was initiated but unsuccessful, and the patient was declared deceased.
Discussion
Rhabdomyosarcoma, which was first described by Raycoff in 1937, is a malignant tumor arising from embryonic mesenchymal cells that can differentiate into skeletal muscle [4]. The literature shows that cardiac sites involved include all heart cahnbers mostly in the the left atrium [5]. Primary malignant tumors of the heart are very rare with frequency of 0.001–0.28% [6]. Cardiac rhabdomyosarcoma accounts for almost 10-20% of all primary malignant neoplasms of the heart [7]. In our case, the lesion in the right heart also led to tricuspid regurgitation, which could contribute to jugular venous distention. Our patient presented with dyspnea and palpitations. Laboratory tests revealed elevated PT and INR values, indicating a coagulopathy, likely secondary to hepatomegaly. The patient, who was followed up in the intensive care unit, died on the second day after surgery due to multiorgan failure secondary to right sided cardiac failure.
Intracardiac rhabdomyosarcoma is a malignancy with an aggressive course and high mortality rate despite all available treatment modalities, such as surgical resection, chemotherapy, and radiotherapy.
- Collucci WS., Shoen FJ., Braunwald E (1997) Primary tumors of the heart. In: Braunwald E, editor. Heart Disease. 5th edn. Philadelphia: WB Saunders Company; 1464-1477.
- Burke AP., Virmani R (1993) Cardiac myxoma. A clinicopathologic study. Am J Clin Pathol 100: 671-680. [Crossref]
- Frank Adusei Poku., Bernice Biney., Samuel Mensah., Sandra Oppong-Twum (2024) Cardiac Rhabdomyosarcoma: An Updated Review of the English Literature From 1980 Through 2023. Circulation 150.
- 4.Kimura A., Tsuji M., Isogai T., Nagata K., Kato K., et al. (2018) A Mass Filling the Right Atrium: Primary Cardiac Rhabdomyosarcoma. Intern Med 3575-3580. [Crossref]
- Kuroda Y., Sadahiro M (2020) Primary Biatrial Cardiac Rhabdomyosarcoma. Braz J Cardiovasc Surg 35: 399-401. [Crossref]
- Holley DG., Martin GR., Brenner JI (1995) Diagnosis and management of fetal cardiac tumors: a multicenter experience and review of published reports. J Am Coll Cardiol 26: 516-520 [Crossref]
- Blondeau P (1996) Primary cardiac tumors – French studies of 533 cases. Thorac Cardiovasc Surg 38:192-195.[Crossref]